


Induction of labour is a critical life-saving intervention that has been proven to have an immeasurable impact on reducing maternal and perinatal mortality and morbidity worldwide.1 Current induction of labour regimes using intravenous oxytocin and prostaglandins, such as dinoprostone, are highly effective but often limited or unavailable in developing countries. dinoprostone, for instance, is widely used in developed countries but its high cost and instability at ambient temperatures limits its usefulness as a cost-effective agent, particularly in developing countries such as Papua New Guinea where the burden of obstetric and perinatal complications are unfortunately the highest in the Pacific region.2
Compared to other prostaglandins, misoprostol has several potential advantages. It is stable at room temperature, inexpensive and can be administered through the oral, vaginal, sublingual and buccal routes. Nevertheless, limited data currently exist concerning the safety, efficacy and feasibility of administering oral misoprostol in routine clinical practice in resource-poor settings.
With the support of a Global Health Research Scholarship provided by the RANZCOG Foundation, we conducted a clinical trial to investigate the safety and efficacy of two different dose regimens of oral misoprostol for induction of labour in Papua New Guinean women.
Background
Prior to the trial, we conducted a pilot observational study to investigate the safety and effectiveness of an oral misoprostol regimen that we have been using in this setting for the past decade.3 Briefly, in this pilot study, more than 6000 labour ward admissions were screened prior to enrolling 209 women who fulfilled the study inclusion criteria and underwent induction of labour. Overall, 74 per cent of the 209 women delivered within 24 hours. Most (90 per cent) delivered vaginally, with 86 per cent having a good outcome for both the mother and baby. Of the 10 per cent who failed induction of labour and underwent caesarean section, a significant proportion of their babies were admitted to special-care nursery compared to babies delivered vaginally. However, their perinatal mortality rate was not significantly higher.4 The only maternal death was not study related and occurred in a patient with post-partum haemorrhage that occurred 15 hours after delivery. Following this study, we concluded that the oral misoprostol regimen for induction of labour, commencing at 25 μg, (Figure 1) is safe, effective and logistically feasible to administer in a resource-limited setting.5
However, because our pilot investigation was an observation study and not a clinical trial, questions related to the oral misoprostol dose escalation nature of our study (Figure 1) as well as the limited safety data of such a regimen and its potential to cause dose-dependent adverse effects still remained. Therefore, we designed the current trial (trial registration ISRCTN10107246) and hypothesised that a regimen commencing with a lower dose of oral misoprostol administered at 12 μg per dose and gradually increased to a maximum of 50 μg per dose over 24 hours will have a non-inferior efficacy and a better safety profile in inducing labour in a developing country setting.
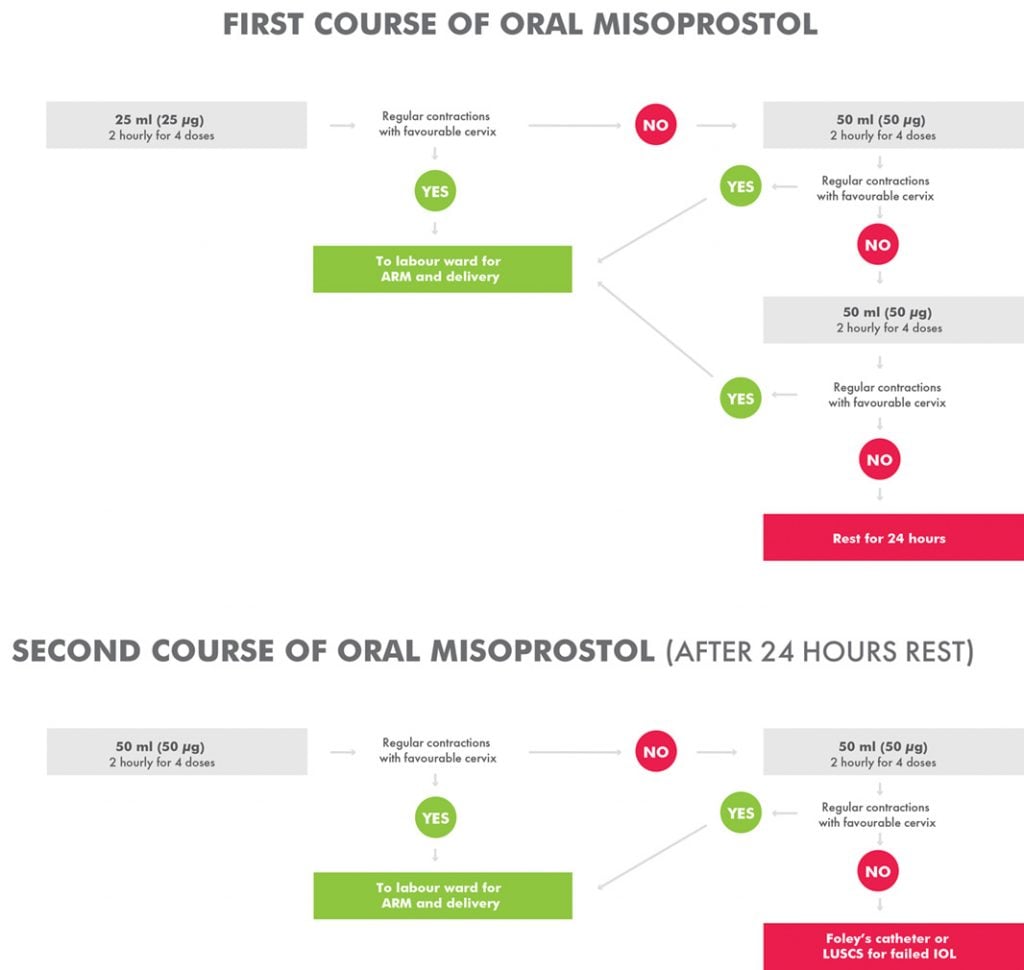
Figure 1. Standard treatment protocol for oral misoprostol induction of labour at Modilon Hospital.
Study setting
The clinical trial was conducted at Modilon General Hospital; a provincial hospital on the northern coast of mainland Papua New Guinea. Modilon hospital has an average of 3000 deliveries per year and an average induction rate of three to five per cent. The hospital has two consultant obstetricians, two registrars and nine midwives who assisted in this study, as well as an operating theatre facility that was used to perform emergency caesarean sections and/or other obstetric and gynaecologial operative procedures.
Study design
After appropriate ethics approvals, trial registration and the formation of a Data Monitoring and Safety Committee, we conducted an open-label randomised controlled trial of a lower dose versus a standard dose of oral misoprostol. Based on computer-generated block randomisation, eligible patients were allocated 1:1 to the standard treatment group commencing at 25 μg (see Figure 1) or a lower dose of oral misoprostol commencing at 12 μg (Figure 2). Allocated treatments were concealed in sealed numbered envelopes that were opened in sequence by study medical or nursing staff and the specified treatment administered.
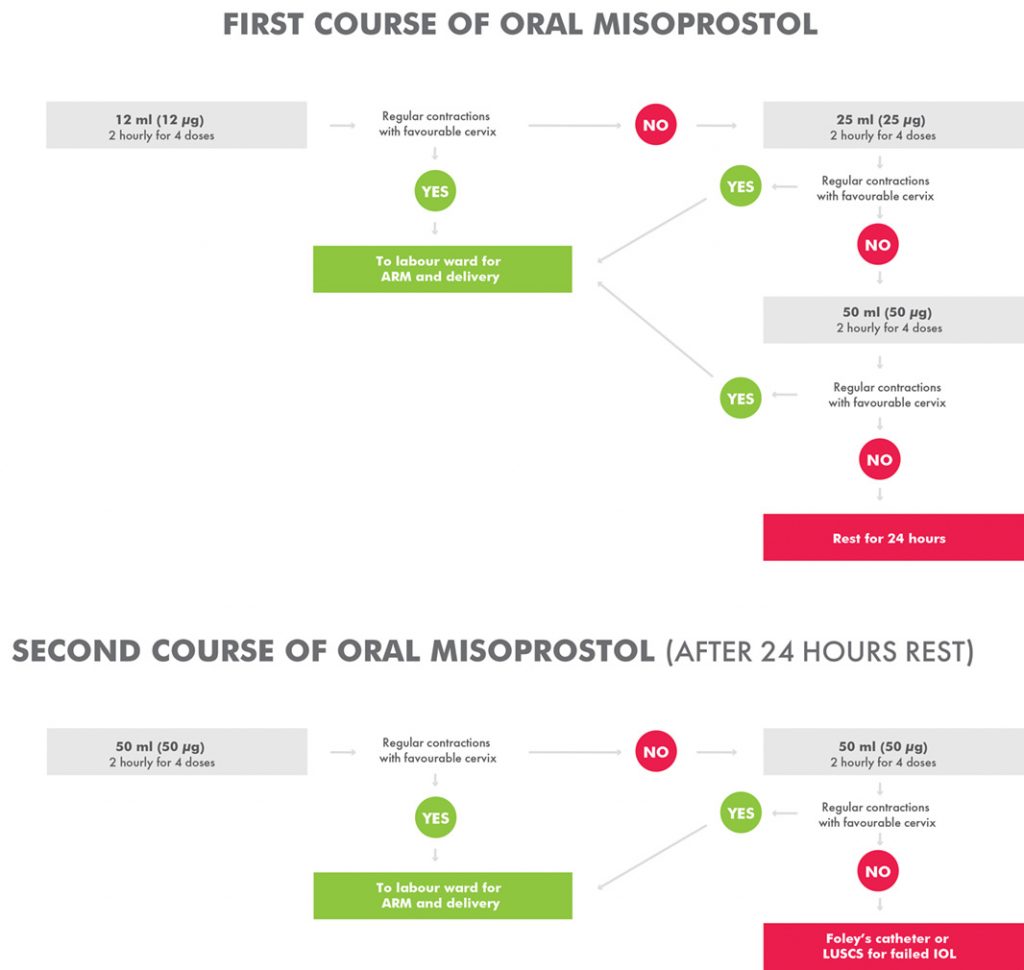
Figure 2. Intervention arm for oral misoprostol induction of labour at Modilon Hospital.
Study endpoints
The primary outcome measured were the proportion of women who had a successful live vaginal delivery without any severe adverse event including: i) failed induction necessitating caesarean section, ii) maternal death, iii) retained placenta, iv) perinatal death, and/or v) neonatal admission to special care nursery. Secondary endpoints included the proportion of i) successful live births delivered vaginally within 24 hours, ii) the proportion of mothers requiring Foley’s catheterisation, iii) the proportion of mothers requiring oxytocin augmentation, iv) neonatal Apgar Scores of seven or below at five minutes post-delivery, and v) reported maternal and neonatal adverse events at four weeks post-discharge.
Women who participated in the study
Women with an indication for induction of labour – such as post-date, pre-labour rupture of membranes, pre-eclampsia – or suspected fetal compromise, such as intrauterine growth restrictions, were considered eligible for enrolment if they fulfilled the study inclusion criteria, which included i) third trimester singleton pregnancies, ii) confirmed cephalic presentations, and iii) a Bishop’s score of less than six.
Oral misoprostol study protocol
A solution of 1 μg/ml was made by dissolving a 200 μg tablet of misoprostol in 200 ml of water. The solution was measured and given in titrated doses as per the study arm requirements. The misoprostol solution was kept at the nurse’s station at room temperature and discarded if not completed within 24 hours. Each dose, either commencing at 12 ml (12 μg/ml) or 25 ml (25 μg/ml) was given at an interval of two hours and doses incremented according to the study arm as outlined in Figures 1 and 2.
Study progress
After the first 60 enrolments (30 participants per arm), an interim analysis was performed that suggested no serious safety concerns and the study was allowed to continue. To date, 256 women have been enrolled as per the study protocol. Follow up at one month post discharge is ongoing, data entry has been completed and analysis of the trial will commence in February of 2019.
Conclusion
With the support of the RANZCOG Foundation Global Health Scholarship, we were able to investigate two oral misoprostol regimens with the aim of reducing maternal and perinatal mortality in our high-burden setting. Despite the simplicity and popularity of oral misoprostol as an induction of labour agent, particularly in developing countries, appropriate evidence is still required to ensure these settings can strive towards reducing the unacceptably high maternal and perinatal mortality rates without increasing the risk of misoprostol-induced adverse events.
References
- WHO. WHO recommendations for induction of labour. Geneva, 2010.
- Bolnga JW, Morris M, Aipit J, Laman M. Maternal and perinatal mortality in resource-limited settings. Lancet Glob Health. 2015;3(11):e672.
- Morris M, Bolnga JW, Verave O, et al. Safety and effectiveness of oral misoprostol for induction of labour in a resource-limited setting: a dose escalation study. BMC Pregnancy Childbirth. 2017;17(1):298. doi: 10.1186/s12884-017-1483-5.
- Morris M, Bolnga JW, Verave O, et al. Safety and effectiveness of oral misoprostol for induction of labour in a resource-limited setting: a dose escalation study. BMC Pregnancy Childbirth. 2017;17(1):298. doi: 10.1186/s12884-017-1483-5.
- Morris M, Bolnga JW, Verave O, et al. Safety and effectiveness of oral misoprostol for induction of labour in a resource-limited setting: a dose escalation study. BMC Pregnancy Childbirth. 2017;17(1):298. doi: 10.1186/s12884-017-1483-5.


Leave a Reply