

The ‘cerebral palsies’ are a diverse group of pathologies with heterogeneous causes. The umbrella clinical term cerebral palsy (CP) is given when a child exhibits non-progressive difficulty in movement control, but it does not signify any particular cause and, until recently, most causes and pathways to CP were unknown. Research into causation has been held back by the long-held and non-evidence-based belief that most cases were owing to acute severe hypoxia at birth. The undefined labels ‘birth asphyxia’ and ‘hypoxic-ischaemic encephalopathy’ are still sometimes given to infants in poor condition at birth when there is no direct evidence of an intrapartum hypoxic sentinel event, a severe metabolic acidosis at birth or brain ischaemia. The condition of these infants at birth sometimes reflects other pathology extending well before labour and possibly back to conception.
Epidemiological research groups, led by Dr Karin Nelson in the USA and Prof Fiona Stanley in Australia in the 1980s, clearly showed most cases of CP were not associated with any putative intrapartum hypoxic event and many cases were associated with clinical risk factors for CP (see Table 1).1 2 3
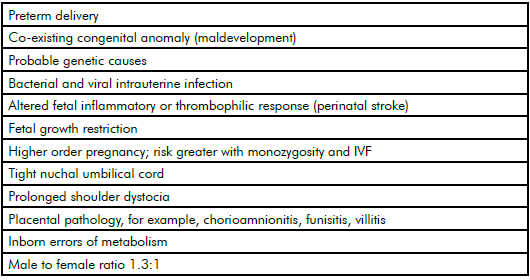
Table 1. Epidemiological risk factors for cerebral palsy.
In later epidemiological studies, our Australian Collaborative Cerebral Palsy Research Group confirmed these risk factors and, in a systematic review of the world literature, showed that neither elective nor emergency caesarean delivery reduced CP risk.4 5 Indeed, a telling statistic is that, over 40 years, the incidence of CP has remained the same at around two per 1000 births, despite the caesarean delivery rate in Australia rising from five to 34 per cent.6 A major component of this rise is defensive obstetrics for fear of CP litigation and the high false-positive rate of intrapartum cardiotocography. Electronic fetal heart rate monitoring has not been shown to reduce the incidence of CP.7
It became clear in the 1980s that fear of CP litigation, the difficulties in using the fetal heart rate as a very poor and indirect surrogate marker of brain health and the rising caesarean rate together were having an untoward effect on obstetrics.8 Since then, four international CP consensus conferences have been convened to help define known clinical risk factors for CP and, in particular, criteria to define the few cases that may have had an acute hypoxic intrapartum cause and timing.9 10 11 12 In many cases, it is easier in retrospect to define that uncommon cause with objective criteria than speculate about the many unknown causes. All obstetricians should gather evidence after a neonate is delivered in potentially poor condition.13 Arterial cord gases, placental pathology and customised weight for gestation to assess potential growth restriction may suggest long-term fetal compromise rather than recent severe hypoxia.14
For decades, we have suspected that some of the cerebral palsies have a genetic basis. In families with an affected singleton child, there is a nine-fold risk of CP in a subsequent sibling and affected parents have a 6.5-fold increased risk of an affected child.15 16 This reflects inherited risk and different causative genes have been found in families with more than one affected member.17 18
Spontaneous de novo mutations causing CP are harder to identify prospectively. Over the last few years, there have been great advances in genetic methodologies that are now uncovering a genomic basis to many neurodevelopmental disorders. CP shares genetic similarities to several neurodevelopmental disorders, including autism, intellectual disability and epilepsy.19 20 21 These are often co-morbidities with CP. Research on these other neurodevelopmental disorders has progressed more quickly than on CP. The Australian Collaborative Cerebral Palsy Research Group has built up a large biobank of DNA from more than 800 CP patients – including 400 trios of affected child, mother and father – linked to an extensive clinical database.
Initially, the methodology of the era was to conduct candidate or genome-wide association studies that look for genetic markers with higher frequency in CP cohorts than in control groups.22This proved a not so suitable methodology, probably because of very high underlying genetic heterogeneity of CP, requiring potentially very large numbers (in 1000s) of CP cases to be investigated. There appears to be a large number of mutations that can disrupt the pathways to brain development or the brain’s defence mechanisms; for example, the fetal inflammatory response or the brain’s defence against thrombosis and fetal stroke.23 24 25 There are a great many putative genetic variants that can directly cause or may contribute to CP through susceptibility triggeredby epigenetic (environmental triggers changing gene function) factors.26 27
Genetic variation can be of different types and can occur in nuclear or in the mitochondrial DNA. Genetic variation affecting larger chunks of DNA is referred to as copy number variation (CNV), while that involving individual nucleotides is referred to as single nucleotide variation (SNV). There is also variation that does not affect the nucleotides, but their modifications; that is, epigenetic variation can affect function (see Figure 1).
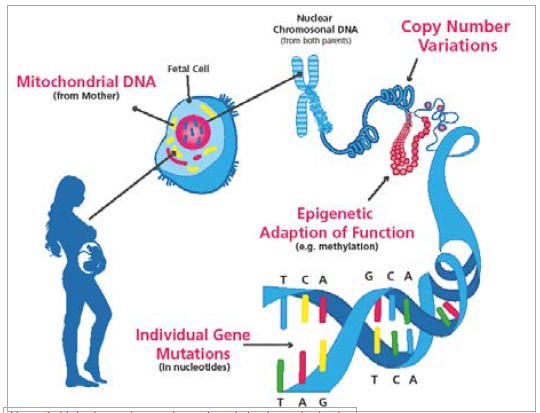
Figure 1. Main sites and types of genetic variation in cerebral palsy.
Two of the main variants are CNVs and SNVs. CNVs consist of submicroscopic structural variations (duplications, deletions or inversions occurring at meiosis) of at least 1kb in size and usually involve many genes. CNVs usually lead to gene dosage imbalance. Single nucleotide variants (SNVs) are a variation in a single or small number of nucleotides. This variation also involves single nucleotide polymorphisms (SNPs); that is, variation found in the populations with a frequency higher that one per cent. Genome-wide CNVs and SNPs can be detected by array-based comparative genomic hybridisation techniques while SNVs – and, in particular, those that are rare (less than 0.1 per cent) or unique – require sophisticated DNA sequencing techniques such as whole exome (WES) and whole genome (WGS) sequencing. Both types of genetic variation can be either inherited or occur de novo in an individual. Most of the CNV, SNP and many SNV variants are benign, unless they happen to hit a developmentally and functionally important area of the genome, in which case they become pathogenic.
In our first such study using WES, of nearly 200 unselected CP cases at least 14 per cent were found to have SNPs with characteristics suggestive of pathogenicity.28 We also have found inherited CNVs in 20 per cent of sporadic CP cases.29 A recent Israeli study has shown that 31 per cent of CP cases had CNVs that were likely to be pathogenic.30 WGS will allow the study of non-coding regions outside the exome that may control gene expression and protein production. Variants affecting movement control may also reside in mitochondrial DNA, which is passed on to the child only from the mother. All these variants require further study. There are different modes of inheritance, variants may have different penetrance. Some disease-causing variants might be X-chromosome linked, which could partly explain the increased burden of CP in hemizygous males.
Genetic research into other, similar and often co-morbid, neurodevelopmental disorders is more advanced than in CP, but similar rates of potentially causative genetic variants have been found (see Table 2).
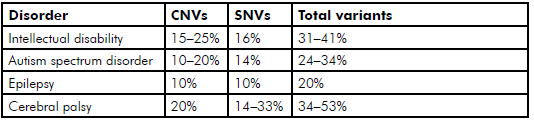
Table 2. Prevalence to date of reported potentially causative genetic variants SNVs and CNVs found through WES and array-comparative genomic hybridisation in neurodevelopmental disorders.
Next-generation sequencing techniques (WES and WGS) reveal many genetic variants in each individual; many will have no pathogenic effect and will be irrelevant to CP causation. The challenge is to find the best and most effective strategies to prioritise the potentially relevant variants according to their genetic characteristics.31 32 The candidate variants found can be classified as likely pathogenic because of their de novo nature, type (stop gain, splice and missense), evolutionary conservation of the mutated residue, known disease association and so forth. These combined characteristics help to prioritise the variants for further investigation.33 However, it is still necessary to demonstrate – using animal, cellular, molecular or in silico models – a functional effect of such a candidate variant in order to resolve its pathogenic (or not) nature. One of the models we are currently exploring is zebrafish, in which we effectively ablate the expression of a specific candidate CP gene and subsequently observe its consequences on fish movement (for example, difficulty swimming away from an adverse stimulus).34 35 Other function studies can be conducted using manipulation of ex vivo cultured primary neurons, for example, mouse hippocampal neurons. Stem cell models also offer a great potential, whether these are patient-derived induced pluripotent stem cells (iPS) or mouse-derived and genetically engineered mouse stem cells.36
Manipulating gene expression can be performed in culture to determine the effect of the predicted causative gene variant on neuron morphology, movement or function. To complement these animal function studies, investigations of global gene expression using RNA sequencing of patient-derived cells allow the study of changes in RNA abundance caused by DNA variants, coding or non-coding. Such transcriptome data (the array of RNA transcripts produced in a particular tissue) also help to identify pathways and networks that might be perturbed37 and further assist in the prioritisation of variants where the functional impact of the mutation may be unclear.
It is likely that, over the coming years, more and more causative genetic variants involved with CP will be found and validated. There are multiple pathways that develop, control and protect movement control and many places that a spontaneous mutation can occur with varying effects on neural function – a great many variants could be involved. Some variants will have a direct effect on movement control, while others will increase susceptibility to CP that may, for example, require an environmental trigger. It is here that the identification of clinical risk factors in epidemiological studies will help direct epigenetic studies. For example, intrauterine infection may, in genetically susceptible fetuses, affect gene expression that controls the normal fetal inflammatory response, either by allowing neurotropic infections to damage the fetal brain or by resulting in a failure to control an exaggerated cytokine response, which also can be neurotoxic.38 39 40 It will take the study of large numbers of cases and their families and much research funding to unravel this complicated story. Genetic factors will certainly not be the only cause of this heterogenetic disorder, but there is a growing understanding of the role they may play.
In the long term, we shall be able to identify common susceptibility genes and gene families leading to CP that can be targeted by personalised therapies. Soon it will be possible to screen the embryo, fetus and newborn for known and validated CP genes, but many genes and genetic variants will still be undiscovered. Again, in the long term, when a known pathogenic gene is identified as early as possible, then gene therapy in the form of gene silencing or gene editing could be possible. It’s been a long road and there is much farther to travel, but now we can see where we should be going.
Funding
These studies have been supported by grants from the Australian NHMRC (1041920, 1019928 and 1099163), The Cerebral Palsy Foundation, The Tenix Foundation, The Women’s & Children’s Hospital Foundation and The Robinson Research Institute Foundation.
Conflicts of interest
The author reports no conflict of interest.
Acknowledgements
The author wishes to thank the Australian Collaborative Cerebral Palsy Research Group.
References
- Nelson KB, Ellenberg JH. Antecedents of cerebral palsy. I. Univariate analysis of risks. Am J Dis Child. 1985; 139:1031-8.
- Nelson KB, Ellenberg JH. Antecedents of cerebral palsy. Multivariate analysis of risk. N Engl J Med. 1986; 315:81-6.
- Blair E, Stanley FJ. Intrapartum asphyxia: a rare cause of cerebral palsy. J Pediatr. 1988; 112:515-19.
- O’Callaghan ME, MacLennan AH, Gibson CS et al. Epidemiologic Associations with Cerebral Palsy. Obstet Gynecol. 2011; 118:576-82.
- O’Callaghan M, MacLennan AH. Caesarean Delivery and Cerebral palsy. A systematic review and meta-analysis. Obstet Gynecol. 2013; 122:1169-75.
- MacLennan AH, Thompson SC, Gecz J. Cerebral Palsy – Causes, pathways, and the role of genetic variants. Am J Obstet Gynecol. 2015; 213:779-88.
- Alfirevic Z, Devane D, Gyte GML. Continuous cardiotocography (CTG) as a form of electronic fetal monitoring (EFM) for fetal assessment during labour. Cochrane Database of Systematic Reviews 2006, Issue 3.Art.No.:CD006066.DOI: 0.1002/14651858.CD006066.
- MacLennan AH, Spencer MK. Projections of Australian obstetricians ceasing practice and the reasons. Med J Aust. 2002; 176: 425-428.
- MacLennan AH for the Australian and New Zealand Perinatal Societies. A Consensus Statement on the Origins of Cerebral Palsy. Med J Aust. 1995;162:85-90.
- MacLennan, AH. A template for defining a causal relation between acute events and cerebral palsy: International consensus statement. BMJ. 1999;319:1054-9.
- Hankins GDV, Speer M. Defining the Pathogenesis and Pathophysiology of Neonatal Encephalopathy and Cerebral Palsy. Obstet Gynecol. 2003;102: 628-36.
- The American College of Obstetricians and Gynecologists and the American Academy of Pediatrics. Neonatal Encephalopathy and Neurologic Outcome. Second Edition. 2014.
- Wong L, MacLennan AH. Gathering the Evidence: Cord gases and placental histology for births with low Apgar scores. Aust N Z J Obstet Gynaecol. 2011; 51: 17-21.
- MacLennan AH, Thompson SC, Gecz J. Cerebral Palsy – Causes, pathways, and the role of genetic variants. Am J Obstet Gynecol. 2015; 213:779-88.
- Tollanes MC, Wilcox AJ, Lie RT, Moster D. Familial risk of cerebral palsy: population based cohort study. BMJ. 349:g4294.
- Hemminki K, Li X, Sundquist K, Sundquist J. High familial risks for cerebral palsy implicate partial heritable aetiology.
Paediatr Perinat Epidemiol. 2007; 21:235-41. - Lerer I, Sagi M, Meiner V et al. Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum Mol Genet. 2005;14:3911-20.
- Lynex CN, Carr IM, Leek JP et al. Homozygosity for a missense mutation in the 67 kDa isoform of glutamate decarboxylase in a family with autosomal recessive spastic cerebral palsy: parallels with Stiff-Person Syndrome and other movement disorders. BMC Neurol. 2004; 30:4:20-9.
- Moreno-De-Luca A, Myers SM, Challman TD et al. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013; 12:406-14.
- Sanders SJ, Murtha MT, Gupta AR et al. De novo mutations revealed by whole exome sequencing are strongly associated with autism. Nature. 2013; 485(7397):237-41.
- Li J, Cai T, Chen H et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NP de novo database. Mol Psych. 2016; 21:290-7.
- O’Callaghan ME, MacLennan AH, Gibson CS, et al for the Australian Collaborative Cerebral Palsy Research Group. Fetal and Maternal Candidate SNP Associations with Cerebral Palsy: A Case-Control Study. Pediatrics. 2012; 129:414-23.
- Bashiri A, Burstein E, Mazor M. Cerebral palsy and fetal inflammatory response syndrome: a review. J Perinat Med. 2006; 34:5-12.
- Gibson CS, MacLennan AH, Goldwater PN et al. The association between inherited cytokine polymorphisms and cerebral palsy. Obstet Gynecol. 2006; 94:674.e1-11.
- Gibson CS, MacLennan AH, Hague WM etal for the South Australian Cerebral Palsy Research Group. Associations between inherited thrombophilias, gestational age, and cerebral palsy. Am J Obstet Gynecol. 2005; 193:1437-43.
- Verkerk AJ, Schot R, Dumee B et al. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. 2009; 85:40-52.
- McMichael G, Bainbridge MN, Haan E, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatr. 2015; 20:176-82.
- McMichael G, Bainbridge MN, Haan E, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatr. 2015; 20:176-82.
- McMichael G, Girirajan S, Moreno-De-Luca A et al. Rare Copy Number Variation in Cerebral Palsy. Eur J Hum Genet. 2013; 22:40-45.
- Segel R, Ben-Pazi H, Zeligson S et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015; 84:1660-8.
- McMichael G, Bainbridge MN, Haan E, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatr. 2015; 20:176-82.
- MacArthur, DG, Manolio, TA, Dimmock, DP et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014; 508: 469-76.
- MacArthur, DG, Manolio, TA, Dimmock, DP et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014; 508: 469-76.
- Nishimura Y, Martin CL, Vazquez-Lopez Aet al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007; 16:1682-98.
- Luo R, Sanders SJ, Tian Y et al. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Amer J Hum Genet. 2012; 91:38-55.
- Goldman SA. Stem and Progenitor Cell-Based Therapy of the Central Nervous System: Hopes, Hype, and Wishful Thinking. Cell Stem Cell. 2016; doi. org/10.1016/j.stem.2016.01.012.
- Voineagu I, Wang X, Johnston P et al.Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011; 474:380-4.
- McMichael G, MacLennan AH, Gibson Get al. Cytomegalovirus and Epstein-Barr virus may be associated with some cases of cerebral palsy. J Matern Fetal Neonatal Med. 2012; 25:2078-81.
- Zhao J, Chen Y, Xu Y, Guanghuan P. Effect of intrauterine infection on brain development and injury. Int J Devl Neuroscience. 2013; 31:543-9.
- Bashiri A, Burstein E, Mazor M. Cerebral palsy and fetal inflammatory response syndrome: a review. J Perinat Med. 2006; 34:5-12.


Leave a Reply