


Knowing the human genome sequence does not automatically mean we have the information required to fully understand human biology and disease.1 Rather, human development and disease are the result of complex processes that emerge and are modified by interactions between an individual’s genotype and the environment. An individual’s genotype is complex and, contrary to popular opinion, is not fixed. Instead, there is considerable variation owing to copy number variation, deletion and insertion within an individual’s non-inherited somatic cells.2 Moreover, a significant contribution to the overall dynamics of an individual’s genome comes from the microbes that make up the microbiome.3 So, how does the environment interact with the genome? Knowing the human genome sequence does not automatically mean we have the information required to fully understand human biology and disease.4 Rather, human development and disease are the result of complex processes that emerge and are modified by interactions between an individual’s genotype and the environment. An individual’s genotype is complex and, contrary to popular opinion, is not fixed. Instead, there is considerable variation owing to copy number variation, deletion and insertion within an individual’s non-inherited somatic cells.5 Moreover, a significant contribution to the overall dynamics of an individual’s genome comes from the microbes that make up the microbiome.6 So, how does the environment interact with the genome?
Environment-genome interactions occur through numerous mechanisms: epigenetic, metabolic or even directly by physical mechanisms7, all of which are modulated by the set of genetic variations present in the individual’s genome. Of course, different combinations of these environment-genome interactions occur over the entire life-course of an individual and while they are important modifiers of disease risk at all stages of life, the signalling that occurs during early development (including pregnancy, birth and early infancy) sets the scene and may be a primary driver of later disease risk.
In the 1940s, Waddington introduced the concept of epigenetics to describe the interaction between the development of a phenotype and the sum of genetic expression and environmental (tissue) interactions.8 Epigenetics, as a term, has since been defined and redefined and has a certain degree of ambiguity associated with it.9 Throughout this article we will discuss epigenetics in terms of changes in gene function that cannot be explained by changes in DNA sequence. Prions and non-coding RNAs are considered to be epigenetic. However, we will focus on post-translational modifications to DNA and histones that cause a change in gene expression and form what is known as the epigenetic code (see Figure 1).10
The epigenetic code is made up of a series of modifications to DNA and proteins related to the control of DNA transcription (for example, histones; see Figure 1) that don’t change the underlying sequence of the DNA, but rather they alter the way a genome functions by changing how accessible it is to the enzymes that are necessary to decode it.11 The action of security guards at a factory can be used as an analogy to illustrate this. Specifically, at the beginning and end of each day, security guards lock and unlock doors in response to signals from their boss (the environment). In this way, they limit the areas of the factory that the workers can access – just like epigenetic marks on the genome. If the security guards are instructed to change their routine then they can alter worker access to store rooms and, in so doing, alter the final products that are produced by the factory.12 Such a change can be positive or negative by promoting the inclusion of inferior parts, or even absence of parts, that limit the lifetime of the final products.
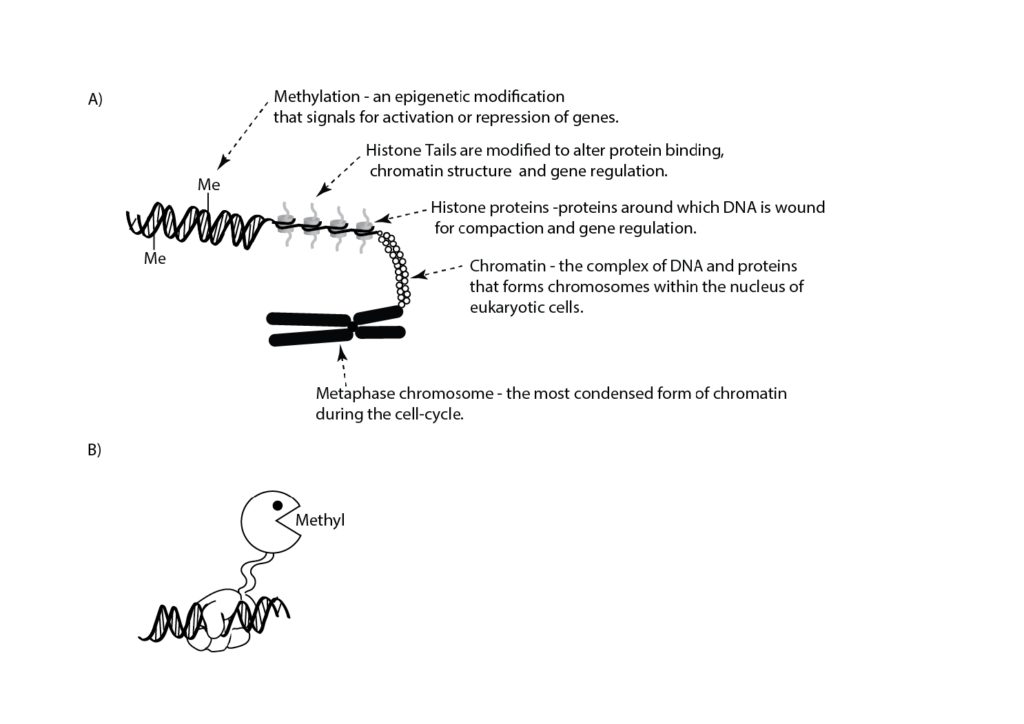
Figure 1. A) Cartoon illustrating features of DNA organisation. An epigenetic modification in the form of methylation (Me) is shown on the double-stranded DNA. Histone tails are subjected to a number of post-translational epigenetic modifications. B) Epigenetic engineering uses composite enzymes that have 1) a DNA binding domain (hand; for example, an enzymatically inactive CAS9 [reviewed in26]) which binds to a specific DNA sequence and 2) an enzyme component (Pac-Man™) that catalyses a specific epigenetic modification (for example, methylation) to turn on or off a specific gene(s).
Epigenetics and the Liggins Institute
Increasing epidemiological and experimental evidence links perinatal factors to later cardiometabolic disease risk – a phenomenon preferentially termed ‘developmental programming’. This is consistent with the evidence that both fixed genomic and epigenetic variation contribute to later disease risk. Despite this, our understanding of the underlying mechanisms remains poorly defined. Data from human cohorts and animal models suggest that epigenetic processes are an important link between the early-life environment, such as maternal diet, and altered metabolism and body composition in offspring in later life.13
There is a strong record of research at the Liggins Institute into the epigenetic processes that underpin developmental programming. As an example, a longstanding and highly productive collaboration between the Liggins Institute and the University of Southampton, UK, demonstrated the importance of the developmental contribution to later adiposity by showing that methylation (see Figure 1) of the retinoid-X receptor-alpha (RXRA) gene at birth was associated with the child’s later adiposity.14Subsequent work has also identified a link between methylation of the RXRA gene at birth and childhood bone mineral content.15 These findings were replicated in independent cohorts, highlighting the robustness of the observations. Interestingly, recent observations have identified a genetic and epigenomic component to post-term birth that may contribute to the increased risks of subsequent life-long complications for these individuals (Prof Wayne Cutfield, Liggins Institute: personal communication). Research at the Liggins Institute has also examined epigenetic profiles in twin births and the potential for epigenetic effects in offspring arising as a result of IVF. Work by Miles et al showed that IVF resulted in altered methylation of genes that appeared to favour childhood growth and metabolism.16 Further, in addition to outcomes linking the early life environment and later obesity and cardiometabolic disorders, work undertaken at the Liggins Institute has also examined effects on later cancer risk and potential mediation by epigenetic processes. Of note, Perry et al showed that similar epigenetic mechanisms underpinned both cancer tumour progression and implantation in human pregnancy.17 Collectively, these studies are consistent with a substantial component of metabolic disease risk having a prenatal developmental basis. Therefore, perinatal epigenetic analysis may have utility in identifying individual vulnerability to later obesity and metabolic disease.
Preclinical models have enormous potential for examining epigenetic mechanisms underpinning developmental programming, transgenerational effects and also opportunities for intervention. Small and large animal models of altered early-life nutrition and direct cell-based platforms have been used by Liggins Institute researchers to characterise a number of epigenetic mechanisms associated with a poor start to life. Rodent models have shown that maternal obesity leads to hypomethylation and enhanced activity of an important regulator of cell growth (in other words, the cell-cycle; P21 gene). Hypomethylation of P21 is present in offspring of obese mothers at the time of birth and likely primes the hepatic dysfunction and later steatosis observed in these animals.18
In sheep, periconceptional undernutrition causes epigenetic changes in offspring in key hypothalamic genes related to energy expenditure (for example, proopiomelanocortin and neuropeptide Y) and glucose homeostasis.19 20 Work by Frank Bloomfield and colleagues has shown that these epigenetic changes are likely to explain the predisposition of these lambs to become obese and suffer from related metabolic disorders.21 22
Changes in developmental trajectory arising from developmental programming were once thought to be permanent. However, research by one of the authors (Mark Vickers) and colleagues was among the first to show that the effects of a poor start to life were indeed reversible when intervention strategies were targeted to particular critical times during development.23 As an example, treatment of neonates with leptin was shown to be able to reverse the adverse metabolic sequalae in rodent offspring following maternal undernutrition. Importantly, the leptin treatment effects highlighted that metabolic plasticity in early development was both sex-specific and directionally dependent upon prior maternal nutritional status.24 25 These observations have now been replicated in a series of independent studies around the world, including in other model species.
Biological data generated by ‘-omics’ platforms are commonly confounded by the nature of sample frequency distributions, which can adversely affect the accuracy of functional inference between biological measure and phenotype.20 Researchers at the Liggins Institute are undertaking different approaches to address this issue. Firstly, Allan Sheppard and colleagues have developed statistical tools that enable improved confidence of functional inference and, in the setting of epigenomic data, also reveal otherwise cryptic information.26 Secondly, novel methods have been developed by one of the authors (Justin O’Sullivan) and colleagues that enable the visualisation of the epigenome in three-dimensions.27 Crucially, this work is identifying spatial associations that help to explain how the information within the genome is accessed to ultimately result in the phenotype that we observe.28
Although most work in the area of epigenetics has focused on changes in DNA methylation, recent work has also examined the role of microRNAs (miRNAs) and histone modifications in gene control. Allan Sheppard and colleagues recently described specific miRNAs associated with steatosis/non-alcoholic fatty liver disease and highlighted biomarkers that may underpin diagnostic profiles and thereby identify at-risk individuals without the need for invasive biopsy, for example, the use of blood plasma.29 Consistent with this, one of the authors (Mark Vickers) and his colleagues have shown that growth hormone treatment of maternal malnutrition-induced hypertension and cardiac hypertrophy in offspring is associated with changes in the Let-7 family of miRNAs.30 Notably, these Let-7 miRNAs have been previously linked to inflammation and cardiovascular development.
Epigenetics and obesity
The role of epigenetics in predisposing individuals to obesity in later life is still being determined. However, work completed to date has a number of important implications. Firstly, the effect sizes associated with epigenetics are considerably greater than those associated with birthweight or maternal body composition.10 This suggests that epigenetic measurements made in the maternal tissues/neonate may be useful predictors of later obesity and other phenotypic outcomes. Secondly, the association between CpG methylation and a child’s later adiposity operates within the normal ranges of maternal nutritional state and birth size. This supports the argument that developmental programming is the consequence of an evolved and potentially adaptive process involving the mechanisms of developmental plasticity.31 Indeed, the data provide strong evidence supporting a role for developmental plasticity in determining individual risk of metabolic disease. Thirdly, the epigenetic data suggest that developmental factors may make a much more significant contribution to phenotypic variation and disease risk than is generally considered. Despite the fact that many of the studies that have been performed to date were limited to identifying correlations and not causative interactions, epigenetic modifications hold great promise across a wide range of metabolic disorders as early biomarkers to predict at-risk individuals.
The future: epigenetics in O&G
The use of drugs that inhibit methyltransferases (for example, azacitidine, decitabine) and histone deacetylases (for example, vorinostat, romidepsin) to modify specific epigenetic marks within the epigenome as therapeutic interventions is already occurring. While these approaches are useful as part of combination therapies, they can have significant side effects as individual treatments. These side effects can be overcome by the development of epigenetic engineering approaches that use new, targeted techniques to manipulate the epigenetic marks at known genes (see Figure 1).32 33 Such approaches have applications across a wide variety of clinical research domains (for example, cancer, protein aggregation diseases, metabolic diseases, neurological and psychiatric diseases) and metabolic diseases where manipulating the expression of a single or small subset of genes can overcome programmed or genetic changes that cause changes to the phenotype of the cell or organism. However, the successful application of epigenetic engineering requires a full understanding of the genome biology in the targeted cells. This understanding must include the inter-relationships between the epigenome and genotype.35 36
Box 1. A glossary of terms
- Copy number variation – the number of copies of a piece of DNA varies from one cell to the next.
Development – the process of growing to maturity. - Developmental programming – a stimulus or insult operating at a critical or sensitive period of development that can result in a longstanding or life-long effect on the structure or function of the organism.
- DNA – a chemical contained in our cells that carries information to build the body. DNA itself has a double helix structure: the information is encoded in the sequence of chemical units along complementary DNA strands. The chemical units are known by the first letters of their names: A, C, G and T. Segments of DNA (genes) act as templates for producing different molecules. DNA information is a unique combination inherited from the mother and father.
- Epigenetics – the study of heritable traits that are not caused by changes in the DNA sequence.
- Epigenome – the spatial arrangement of the total set of all the epigenetic marks that are present in a genome.
- Epigenetic code – a code consisting of epigenetic marks that is dictated in response to environmental signals.
- Epigenetic engineering – targeted epigenetic modification of specific genes.
- Epigenetic marks – there are three basic classes of epigenetic marks:
- post-translational modifications that occur on the proteins around which the DNA is wrapped (histones);
- methylation of the deoxyribonucleotide bases within DNA; and
- hydroxymethylation of DNA.
- Gene – segments of DNA that encode the instructions to build molecules (usually proteins) that make our cells and keep them functioning.
- Genome – all of the genes or genetic material present in cell or organism.
- Genotype – the set of genetic variations present in the individual’s genome. This includes all the changes from the reference genome that are unique to the individual.
- Histones – a group of proteins that form a complex (the nucleosome) about which DNA is wound inside cells with nuclei.
- Histone deacetylases – these remove acetyl marks (a post-translational modification) from histone proteins.
- Hypomethylation – a measurable reduction in the amount of methylation at a site in the DNA or histones.
- Leptin – a ‘satiety’ hormone made primarily by fat cells that helps regulate energy balance.
- Methylation – an epigenetic modification that involves the addition of a methyl group to the DNA or histone.
- Methyltransferases – enzymes that add methyl groups to their target substrate (for example, DNA or histone proteins).
- Microbiome – the collection of micro-organisms present on/in an individual.
- microRNAs (miRNA) – a group of small RNAs that do not code for proteins, but act to regulate genes and gene expression.
- Nucleotide – the chemical units of DNA, known by the first letters of their names: A, C, G and T. Each nucleotide is made up of a nitrogenous base (for example, adenine, thymine, guanine or cytosine) together with sugar and phosphate groups. The order of As, Cs, Ts and Gs along a DNA strand encodes information; segments of DNA act as templates for producing different molecules.
- Somatic cell – any non-inherited, non-reproductive cell in an organism.
- Undernutrition – a deficiency of one or more essential nutrients or calories.
- Variant – a DNA difference or change on a continuum from benign to pathogenic. Variants can range from large rearrangements, deletions or duplications of massive segments of DNA (copy number variants, or CNVs) right down to single nucleotide changes (single nucleotide variants, or SNVs).
References
- Green ED. The Genomic Landscape circa 2014 [Internet]. www.genome.gov/pages/research/intramuralresearch/dircalendar/ctga2014/ctga2014_lec01_color.pdf.
- O’Huallachain M, Karczewski KJ, Weissman SM et al. Extensive genetic variation in somatic human tissues. Proc Natl Acad Sci USA. 2012; 109(44):18018-23.
- Grice E, Segre J. The human microbiome: our second genome. Annu Rev Genomics Hum Genet. 2012; 13:151-70.
- Green ED. The Genomic Landscape circa 2014 [Internet]. www.genome.gov/pages/research/intramuralresearch/dircalendar/ctga2014/ctga2014_lec01_color.pdf.
- O’Huallachain M, Karczewski KJ, Weissman SM et al. Extensive genetic variation in somatic human tissues. Proc Natl Acad Sci USA. 2012; 109(44):18018-23.
- Grice E, Segre J. The human microbiome: our second genome. Annu Rev Genomics Hum Genet. 2012; 13:151-70.
- Sontam DM, Firth EC, Tsai P et al. Different exercise modalities have distinct effects on the integrin-linked kinase (ILK) and Ca2+ signaling pathways in the male rat bone. Physiol Rep. 2015; 3(10):e12568.
- Bard JBL. Waddington’s Legacy to Developmental and Theoretical Biology. Biol Theory. 2008; 3(3):188-97.
- Ptashne M. Epigenetics: core misconcept. Proc Natl Acad Sci USA. 2013 Apr 30 [cited 2014 Apr 29]; 110(18):7101-3.
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001; 293(5532):1074-80.
- Ptashne M. Epigenetics: core misconcept. Proc Natl Acad Sci USA. 2013 Apr 30 [cited 2014 Apr 29]; 110(18):7101-3.
- Schierding WS, Vickers M, O’Sullivan JM, Cutfield WS. Epigenetics. In: Polin RA, Benitz WE, Abman SH, editors. Fetal and Neonatal physiology. 5th ed. Elsevier Clinical Solutions, Elsevier; 2016.
- Cutfield WS, Hofman PL, Mitchell M, Morison IM. Could epigenetics play a role in the developmental origins of health and disease? Pediatr Res. 2007; 61(5 PART 2 SUPPL.):68-75.
- Godfrey KM, Sheppard A, Gluckman PD et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011; 60(5):1528-34.
- Harvey NC, Sheppard A, Godfrey KM et al. Childhood bone mineral content is associated with methylation status of the RXRA promoter at birth. J Bone Miner Res. 2014; 29(3):600-7.
- Miles HL, Hofman PL, Peek J et al. In vitro fertilization improves childhood growth and metabolism. J Clin Endocrinol Metab. 2007; 92(9):3441-5.
- Perry JK, Lins RJ, Lobie PE, Mitchell MD. Regulation of invasive growth: similar epigenetic mechanisms underpin tumour progression and implantation in human pregnancy. Clin Sci (Lond). 2010; 118:451-7.
- Dudley KJ, Sloboda DM, Connor KL et al. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 6(7): e21662. doi:10.1371/journal.pone.0021662.
- Stevens A, Begum G, Cook A et al. Epigenetic changes in the hypothalamic proopiomelanocortin and glucocorticoid receptor genes in the ovine fetus after periconceptional undernutrition. Endocrinology. 2010; 151(8):3652-64.
- Begum G, Davies A, Stevens A et al. Maternal undernutrition programs tissue-specific epigenetic changes in the glucocorticoid receptor in adult offspring. Endocrinology. 2013; 154(12):4560-9.
- Stevens A, Begum G, Cook A et al. Epigenetic changes in the hypothalamic proopiomelanocortin and glucocorticoid receptor genes in the ovine fetus after periconceptional undernutrition. Endocrinology. 2010; 151(8):3652-64.
- Begum G, Davies A, Stevens A et al. Maternal undernutrition programs tissue-specific epigenetic changes in the glucocorticoid receptor in adult offspring. Endocrinology. 2013; 154(12):4560-9.
- Vickers MH, Gluckman PD, Coveny AH et al. Neonatal leptin treatment reverses developmental programming. Endocrinology. 2005; 146(10):4211-6.
- Vickers MH, Gluckman PD, Coveny AH et al. The effect of neonatal leptin treatment on postnatal weight gain in male rats is dependent on maternal nutritional status during pregnancy. Endocrinology. 2008; 149(4):1906-13.
- Gluckman PD, Lillycrop K, Vickers MH et al. Metabolic plasticity during mammalian development is directionally dependent on early nutritional status. Proc Natl Acad Sci USA. 2007; 104(31):12796-800.
- Pleasants AB, Wake GC, Shorten PR et al. A new, improved and generalizable approach for the analysis of biological data generated by -omic platforms. J Dev Orig Heal Dis. 2015; 6(1):17-26.
- Pichugina T, Sugawara T, Kaykov A et al. A diffusion model for the coordination of DNA replication in Schizosaccharomyces pombe. Sci Rep. 2016 Jan 5; 6:18757.
- Schierding W, Antony J, Cutfield WS, Horsfield JA, O’Sullivan JM. (In review) Intergenic GWAS SNPs are key components of the spatial and regulatory network for human growth. Hum Mol Genet.
- Zarfeshani A, Ngo S, Sheppard A. MicroRNA Expression Relating to Dietary-Induced Liver Steatosis and NASH. J Clin Med. 2015; 4(12):1938-50.
- Gray C, Li M, Patel R, Reynolds CM, Vickers MH. Let-7 miRNA profiles are associated with the reversal of left ventricular hypertrophy and hypertension in adult male offspring from mothers undernourished during pregnancy after preweaning growth hormone treatment. Endocrinology. 2014; 155(12):4808-17.
- Gluckman PD, Hanson MA, Beedle AS. Non-genomic transgenerational inheritance of disease risk. BioEssays. 2007; 29(2):145-54.
- Kungulovski G, Jeltsch A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2015; 32(2):101-13.
- Falahi F, Sgro A, Blancafort P. Epigenome Engineering in Cancer: Fairytale or a Realistic Path to the Clinic? Front Oncol. 2015; 5:22.
- Pichugina T, Sugawara T, Kaykov A et al. A diffusion model for the coordination of DNA replication in Schizosaccharomyces pombe. Sci Rep. 2016 Jan 5; 6:18757. 34Schierding W, Antony J, Cutfield WS, Horsfield JA, O’Sullivan JM. (In review) Intergenic GWAS SNPs are key components of the spatial and regulatory network for human growth. Hum Mol Genet.
- Schierding W, Cutfield WS, O’Sullivan JM. The missing story behind Genome Wide Association Studies: single nucleotide polymorphisms in gene deserts have a story to tell. Front Genet. 2014 Jan [cited 2014 Nov 18]; 5:39.


Leave a Reply